If this is your first time using this app, consider taking the guided interactive tour by clicking the orange button in the top right. If you need a hint about what a specific setting or button does, click the question mark in the top right corner, this will display content-aware hints.
If that does not answer all of your questions, you can find a complete documentation below.
You can address any further questions about WEADE to Nils Trost, nils.trost@stud.uni-heidelberg.de
Your data is only stored during the session, it is not accessible for anyone else but you for the analyses. Please refer to our Terms of Use for more details.
If the application shows a loading screen for more than roughly 5 minutes, something most certainly went wrong. Try reloading the page and running the analysis again. If the issue keeps happening, contact Nils Trost, nils.trost@stud.uni-heidelberg.de.
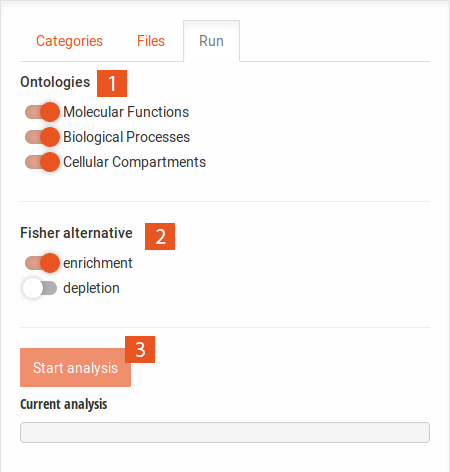
The tool uses the Fisher's exact test to calculate the enrichment of a category or a term from a list of genes. The resulting p-Values are adjusted for multiple hypothesis testing with the Benjamini-Hochberg method when testing on categories. Additionally, the term-frequency or odds ratio is calculated.
The tool performs a Mann-Whitney U-test on a set of genes with an associated continuous measure. The resulting p-Values are adjusted for multiple hypothesis testing with the Benjamini-Hochberg method when testing on categories. This way, no threshold has to be set, all genes are considered and weighted according to the provided measure. Additionally, the delta-rank of each category or term is calculated.
The side tabs contain the options that you can set to perform the analyses.
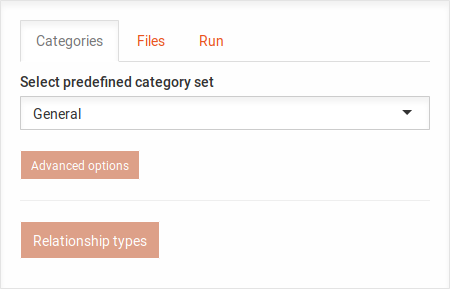
In the Categories tab, you can select the predefined category sets for the analysis. You may click on the Advanced options or the Relationship types button to reveal the following options:
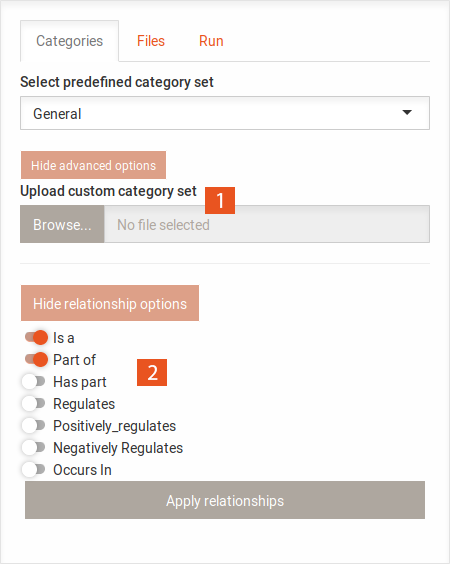
GO id GO term Name for category
Changing the set of categories will reset the heat map and history, as it makes the analyses incomparable.
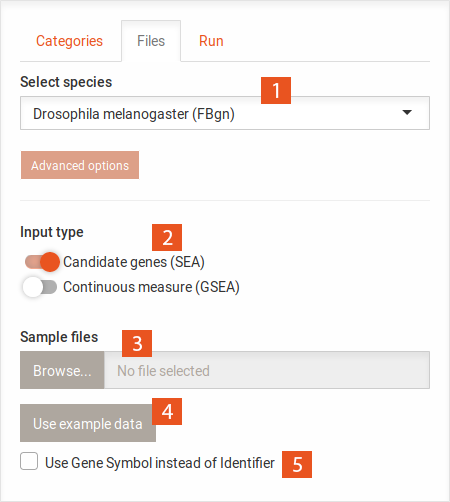
...
Gene1
Gene2
Gene3
...
...
Gene1,-0.2
Gene2,2.3
Gene3,1.4
...
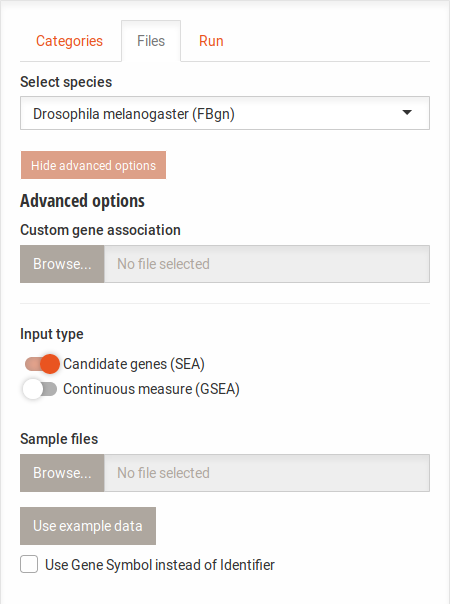
Clicking the
Advanced options button below the species selection shows the upload area for custom annotation files. With
this, you can use an alternate identifier for species that are present or run the analysis on other species. The file
format for this is as follows:
...
FBgn0041711 GO:0005576
FBgn0041711 GO:0048067
FBgn0041711 GO:0016853
FBgn0041711 GO:0042438
FBgn0042110 GO:0003674
FBgn0042110 GO:0003824
FBgn0042110 GO:0016740
FBgn0042110 GO:0016772
FBgn0037149 GO:0005575
FBgn0037149 GO:0003674
FBgn0037149 GO:0008150
FBgn0039007 GO:0007165
FBgn0039007 GO:0007154
FBgn0039007 GO:0007186
FBgn0039007 GO:0008150
...
The file should be a tab separated text file with two columns. The first column should contain the gene identifiers
and the second column the corresponding GO term. If a gene identifier is associated with more than one GO term, put each
association in a separate line.
You may also add a third column containing gene symbols for display instead of the gene identifiers.
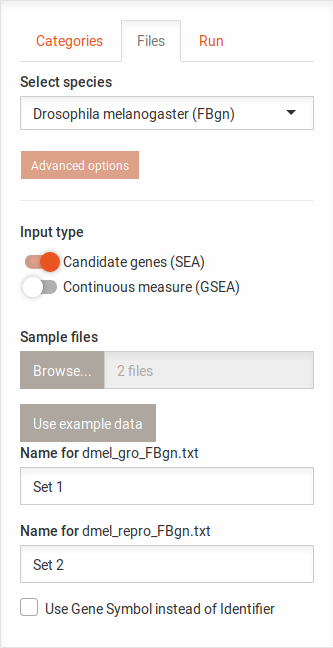
After uploading input files, you can enter names for the sets. These names will be displayed in the Venn diagram. This optional but helpful, if the file names are not clear.
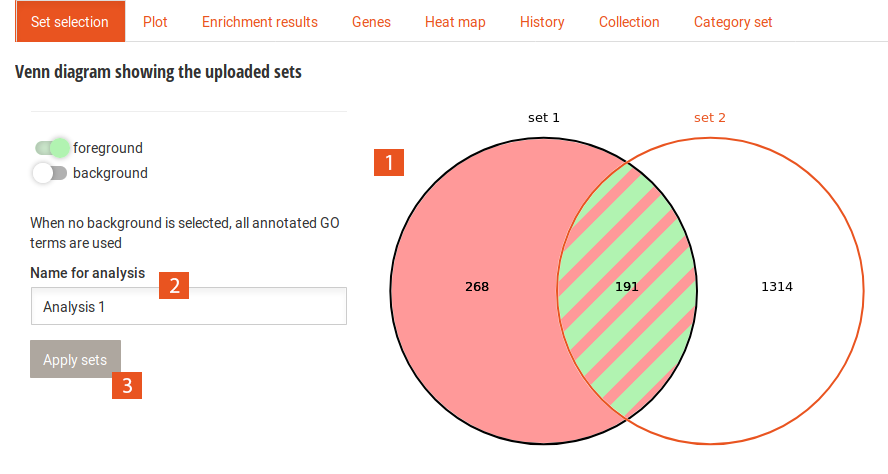
The main tabs display the set selection Venn diagram as well as the results of the analyses.
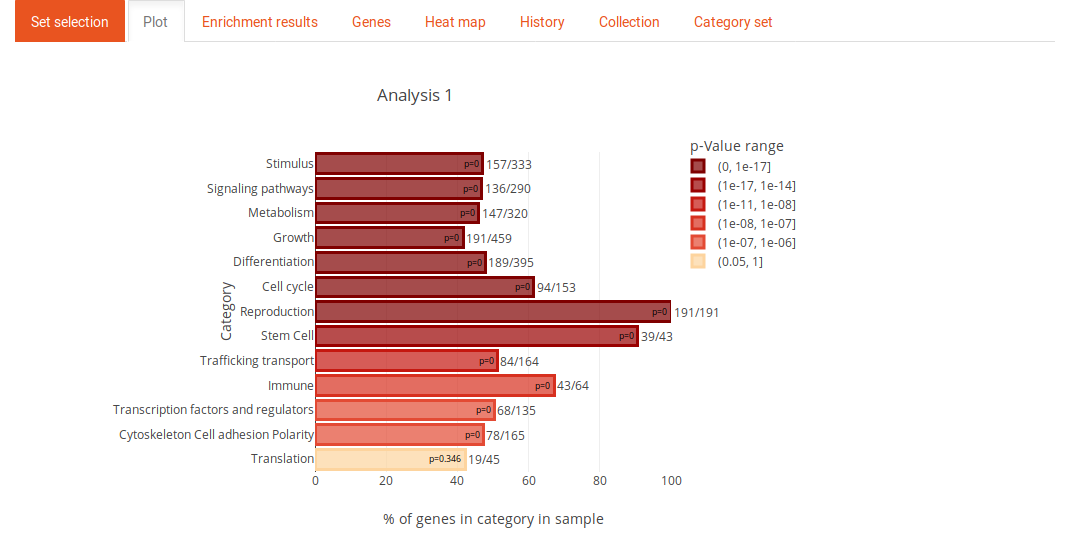
The plot tab contains a plot of the enrichment results. If a list of significant genes was used for the analysis (SEA), the length of the bars is the odds ratio (term-frequency). If the analysis was performed on a list of genes with a continuos measure (GSEA), the length of the bars corresponds to the delta rank of the MWU test. The color of the bars represents the p-Value.
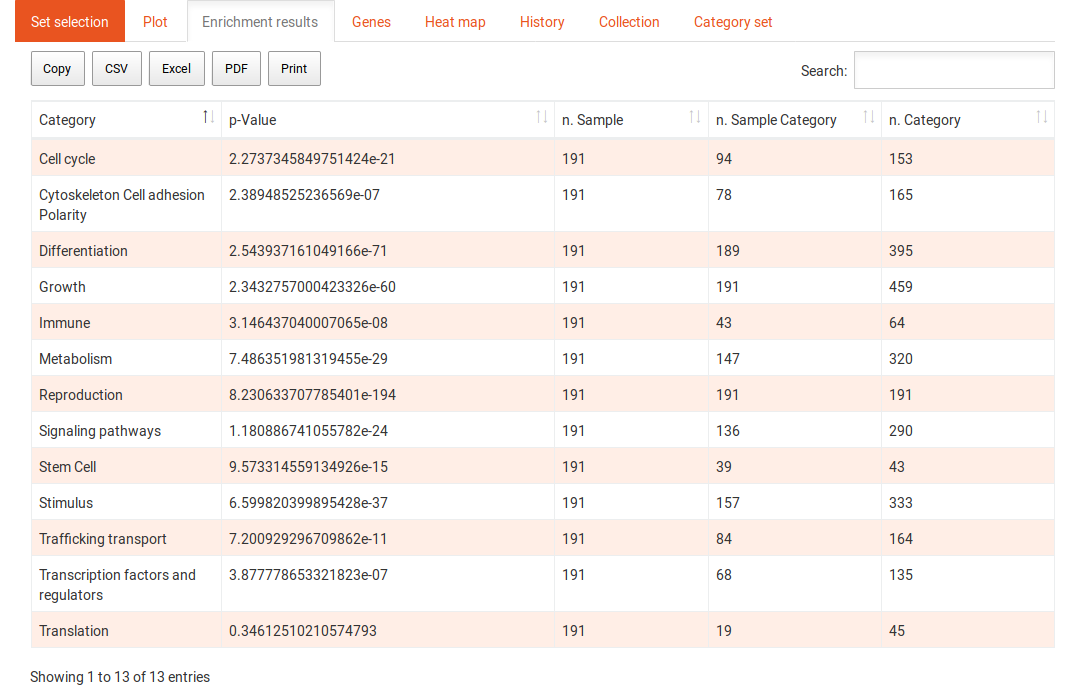
The enrichment results tab shows the results in a tabular format. The first column contains the names of the categories, the second contains the p-values of the enrichment of those categories. The last three columns contain the numbers of genes in the sample, in the sample and in the category, and in the background and in the category, respectively, for SEA and the delta-rank for GSEA. The table can be sorted and filtered with the search and the results can be downloaded in several formats. Clicking on one of the categories shows the following options:
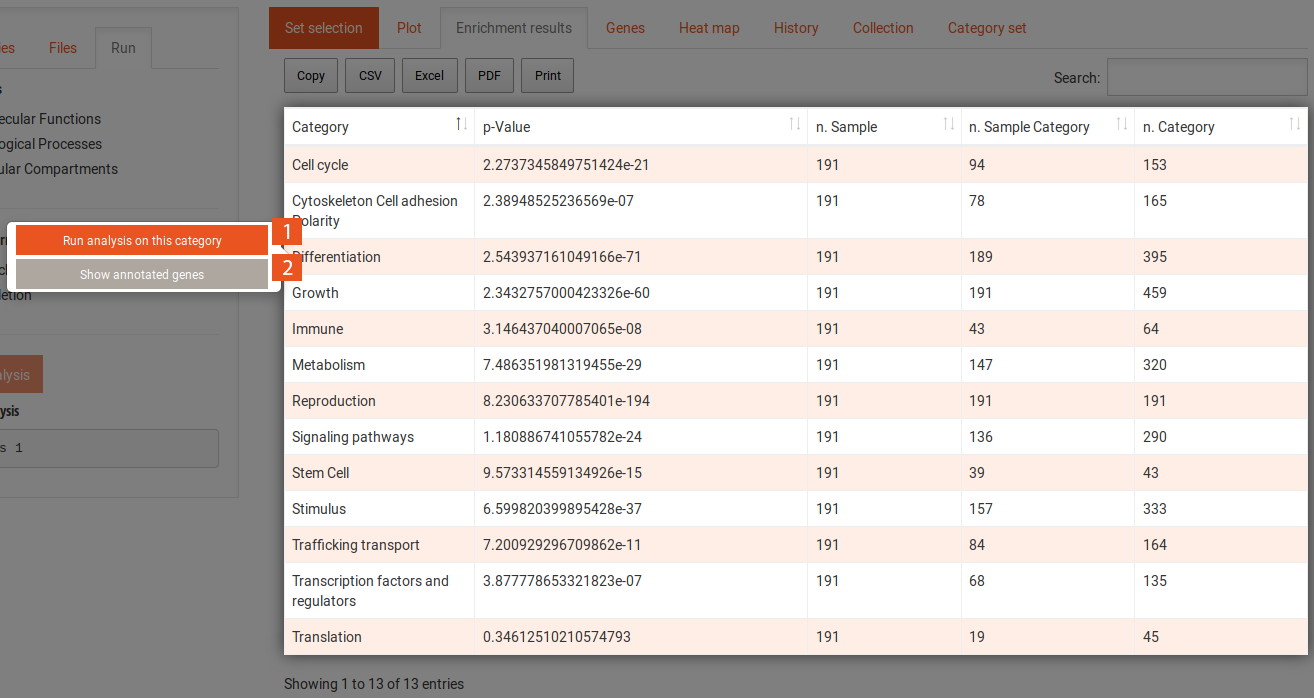
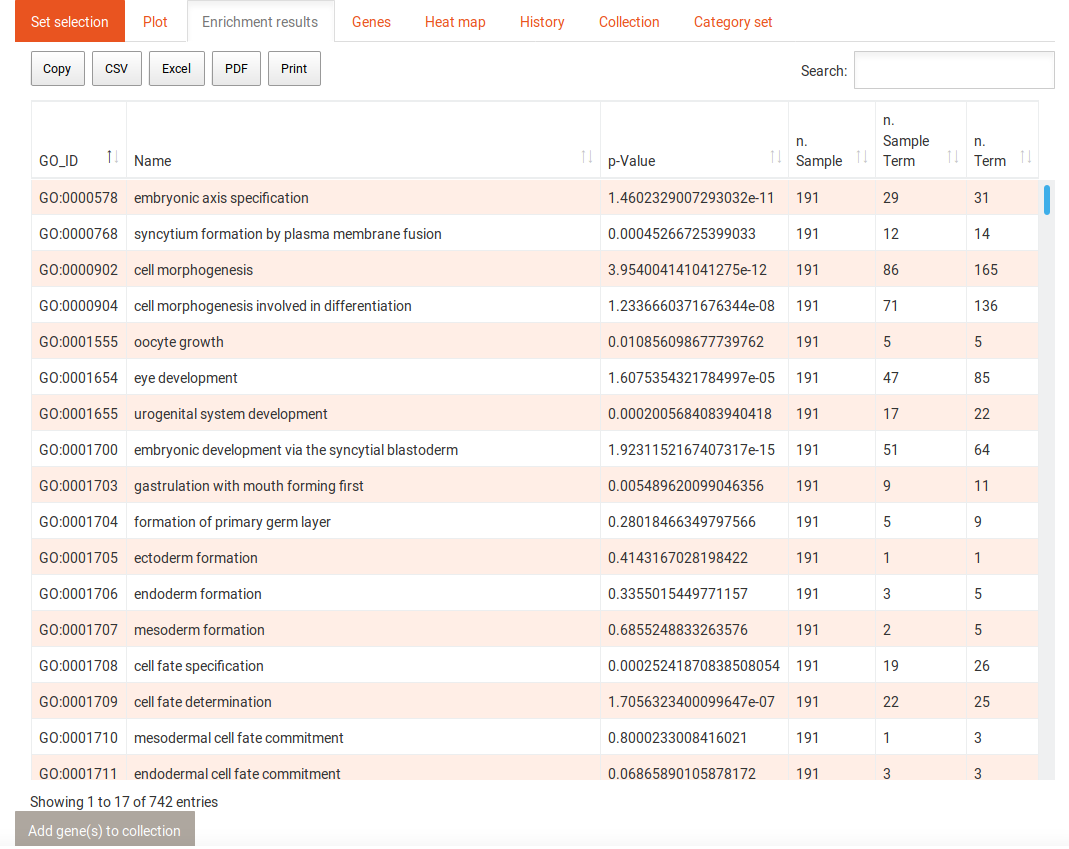
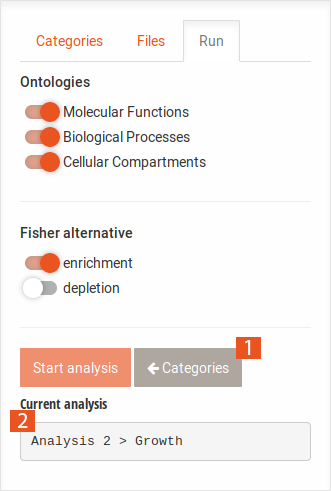
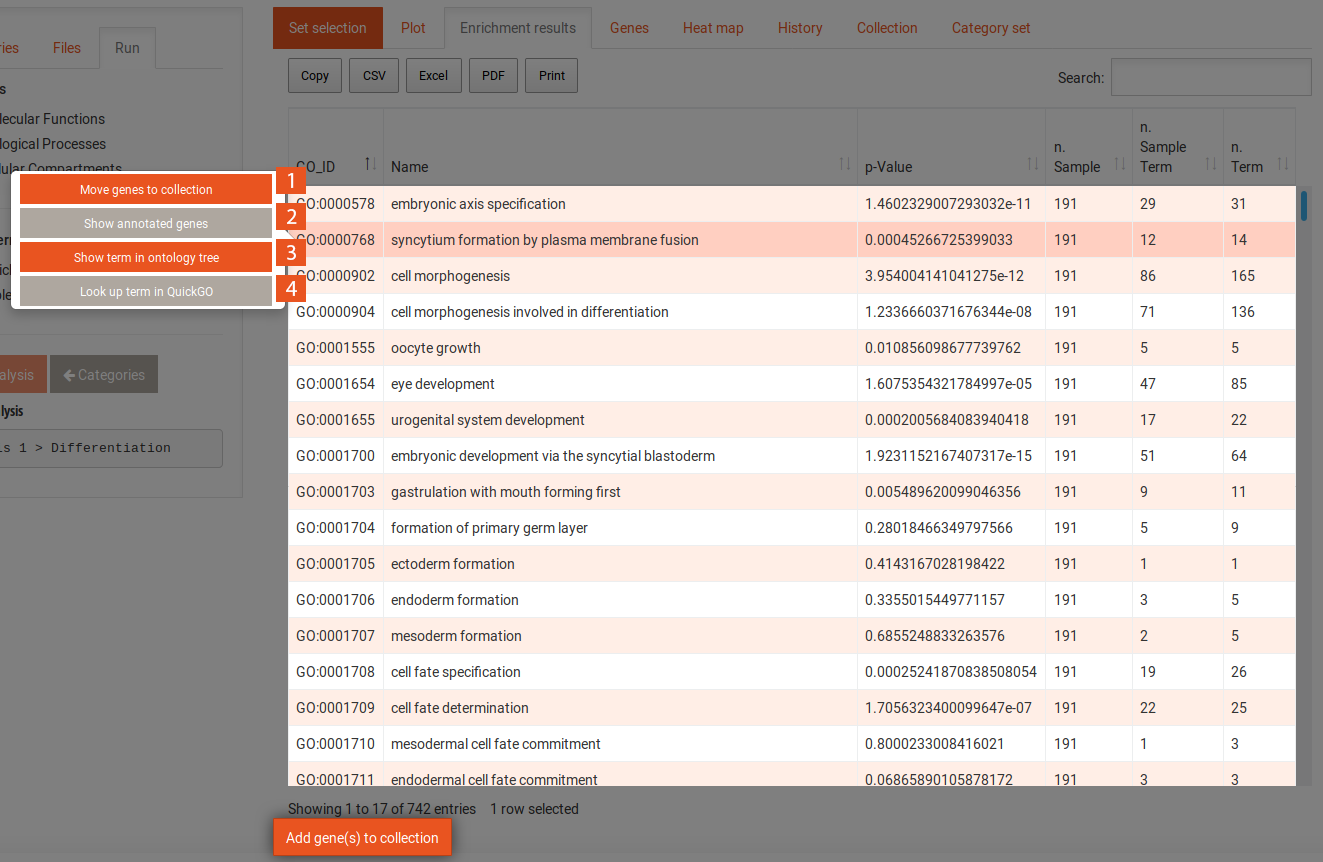
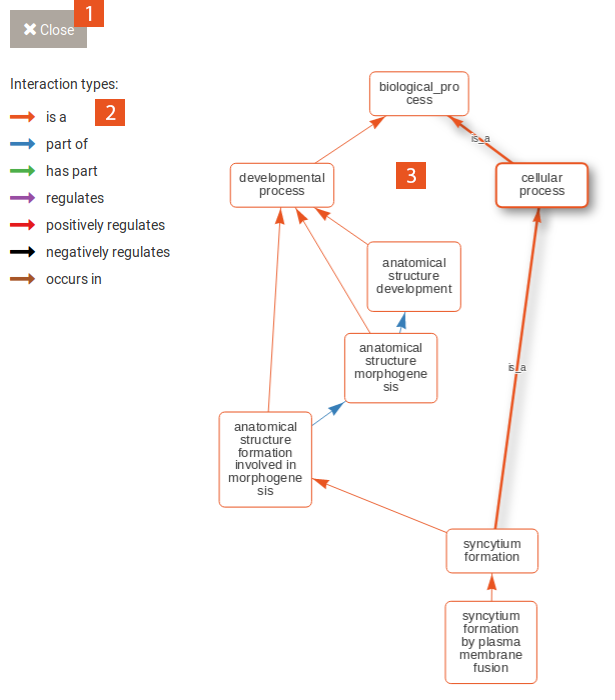
When you click on the Run analysis on this category button of a selected category, the enrichment results table will change to now include the terms that contribute to the enrichment of the category. You can use the Back to categories button in the Run tab to go back to the previous view of all categories. Selecting a term in this table reveals the following options:
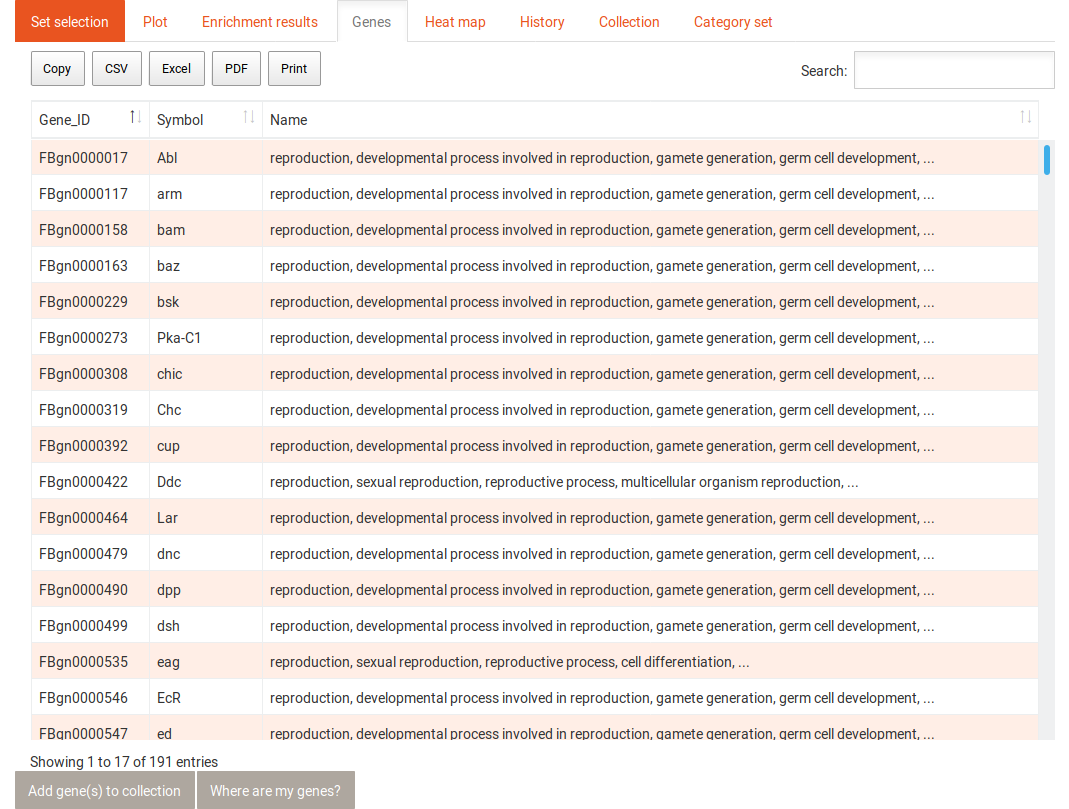
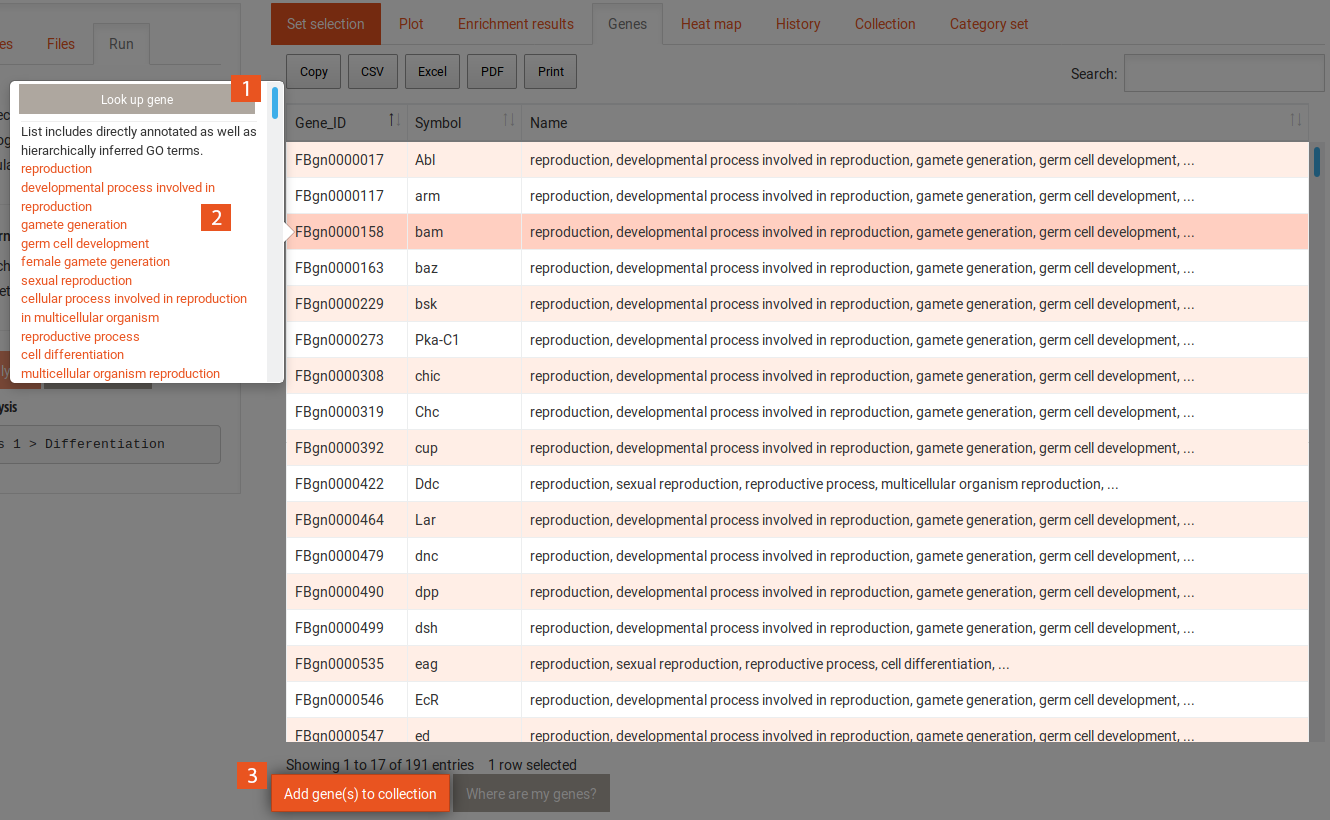
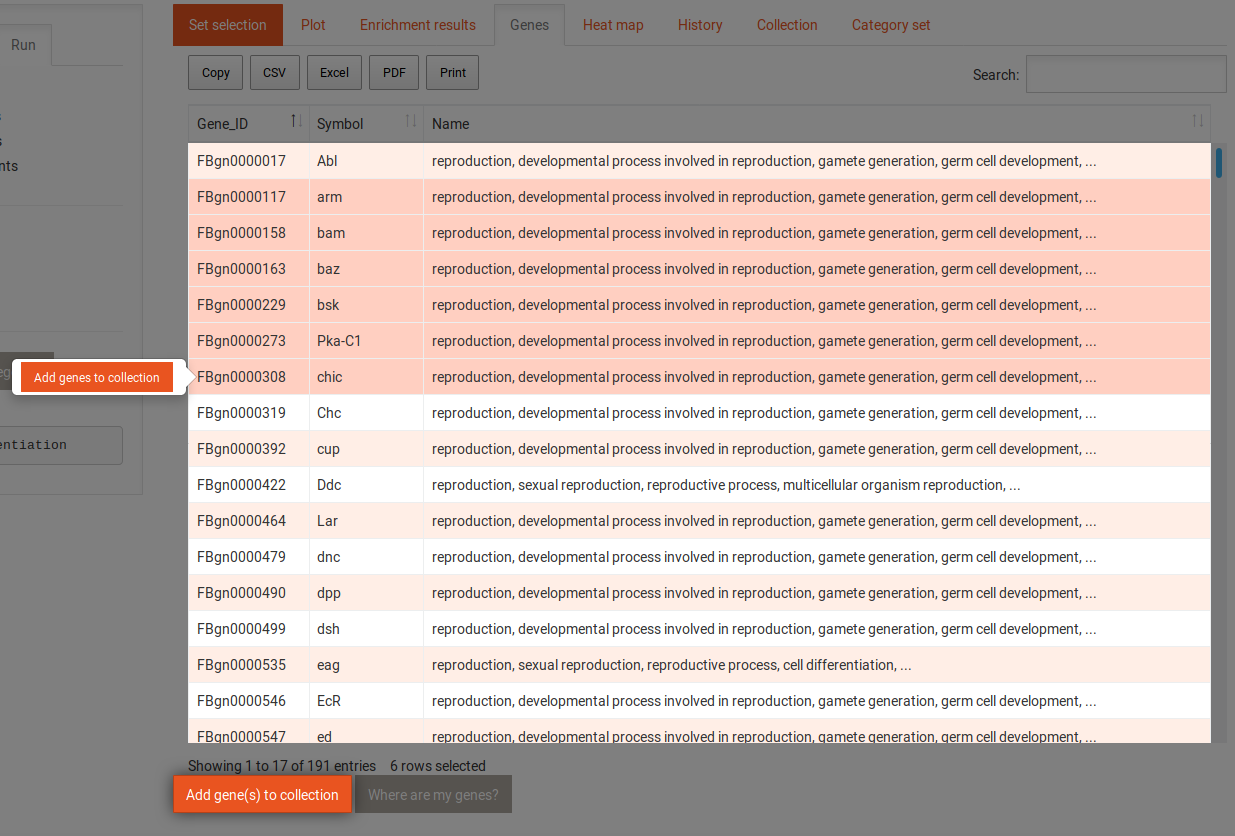
The gene tab contains a table with all genes and the category or term to which they were annotated. This table can also be sorted and filtered and its content be downloaded in several formats. Selecting one gene in this table shows the following information:
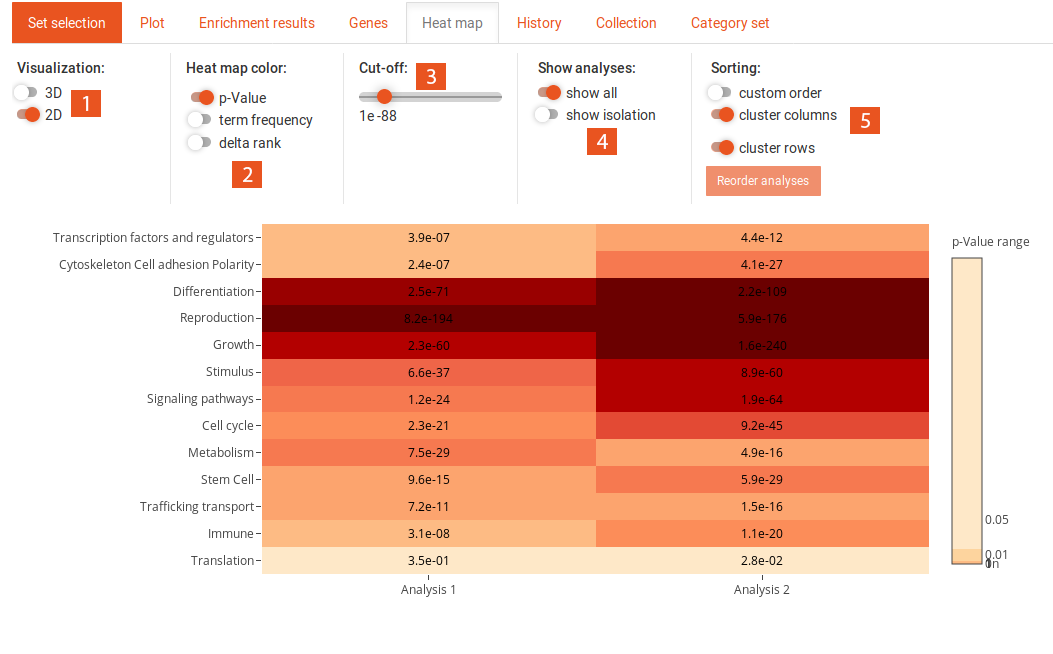
This tab shows a heat map of previous analyses, if at least two of the same type (SEA/GSEA) have been performed. The options for the heat map can be found above.
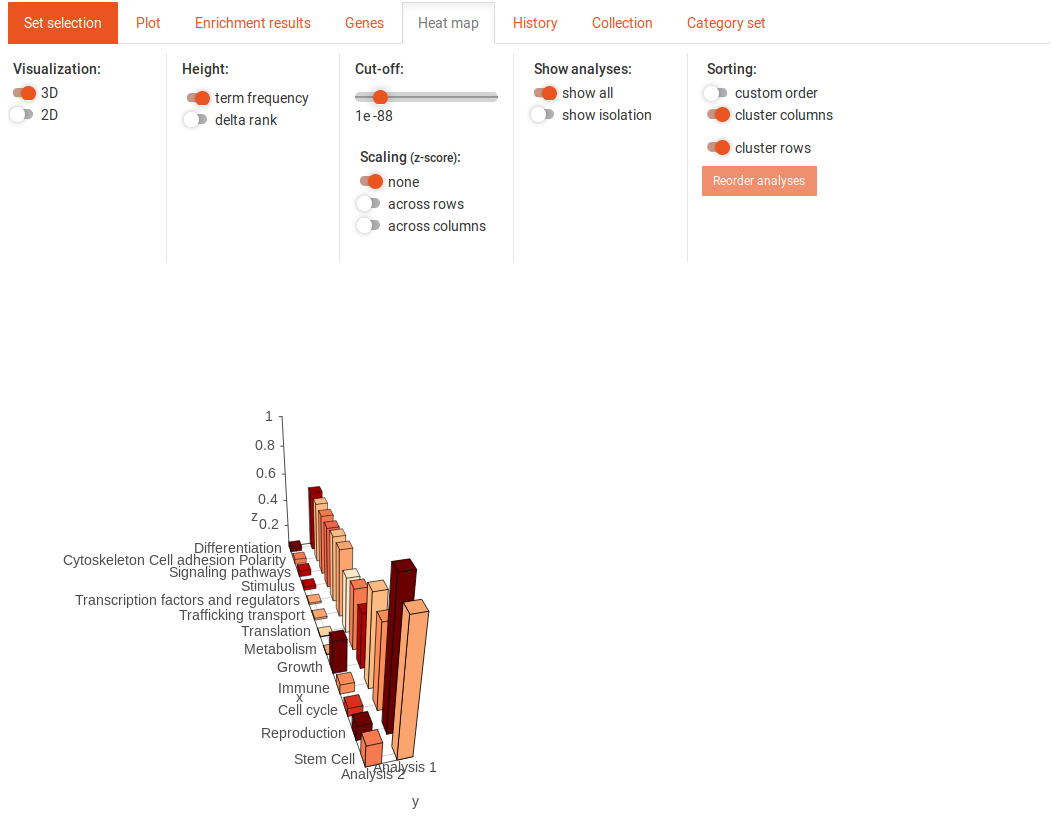
This image shows the 3D heat map. You can move the view around by clicking on it and dragging. In the 3D heat map, the colour always corresponds to the p-value. You can adjust the colour cut-off in the same way as in the 2D heat map. The height of the bars represent either the term-frequency (SEA) or the delta-rank (GSEA). You can display either the absolute values of the term-frequency and delta-rank or use scaling with the z-score. This scaling can be done across columns or across rows.
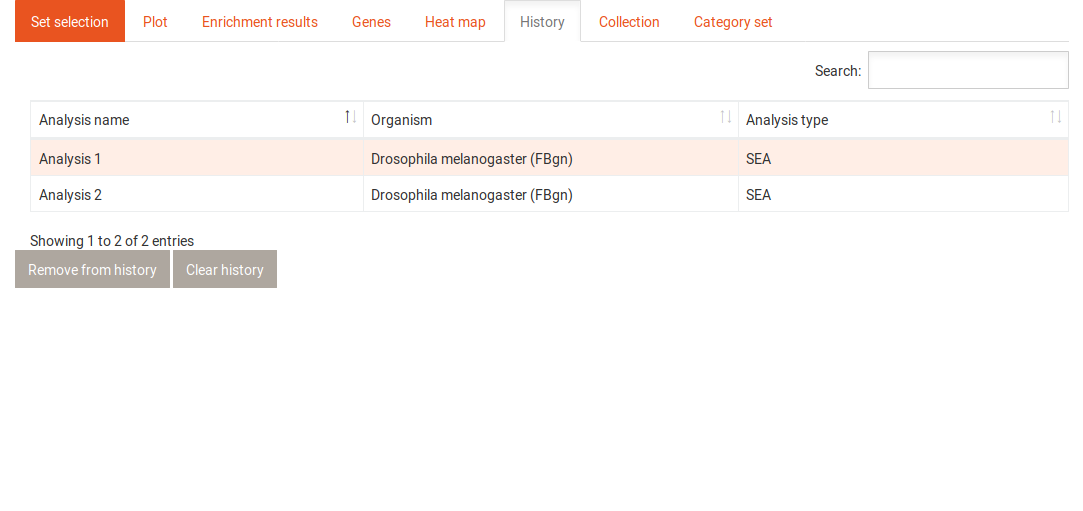
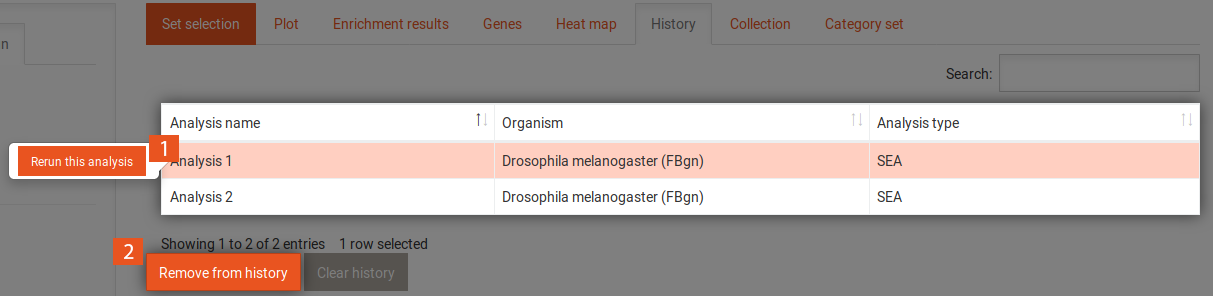
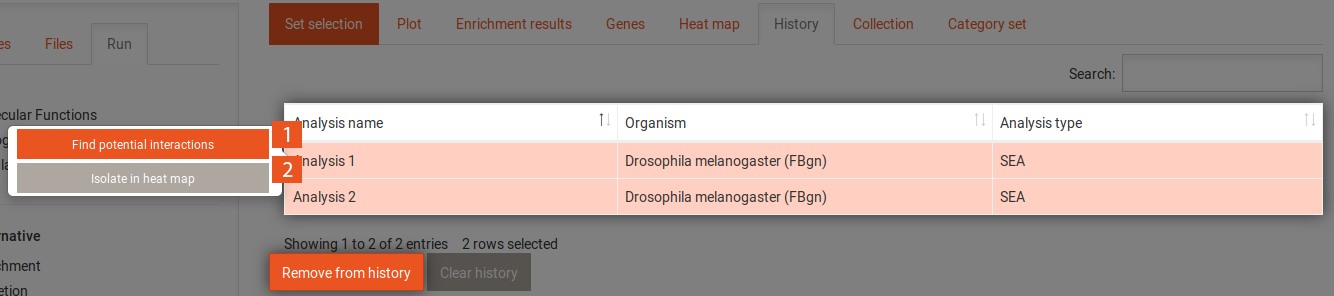
The history tab contains a table with all previously performed analyses. It shows the name of each analysis, the organism that was used and the type of analysis (SEA/GSEA). Clicking on one of the analyses gives you access to the following options:
Clicking on the
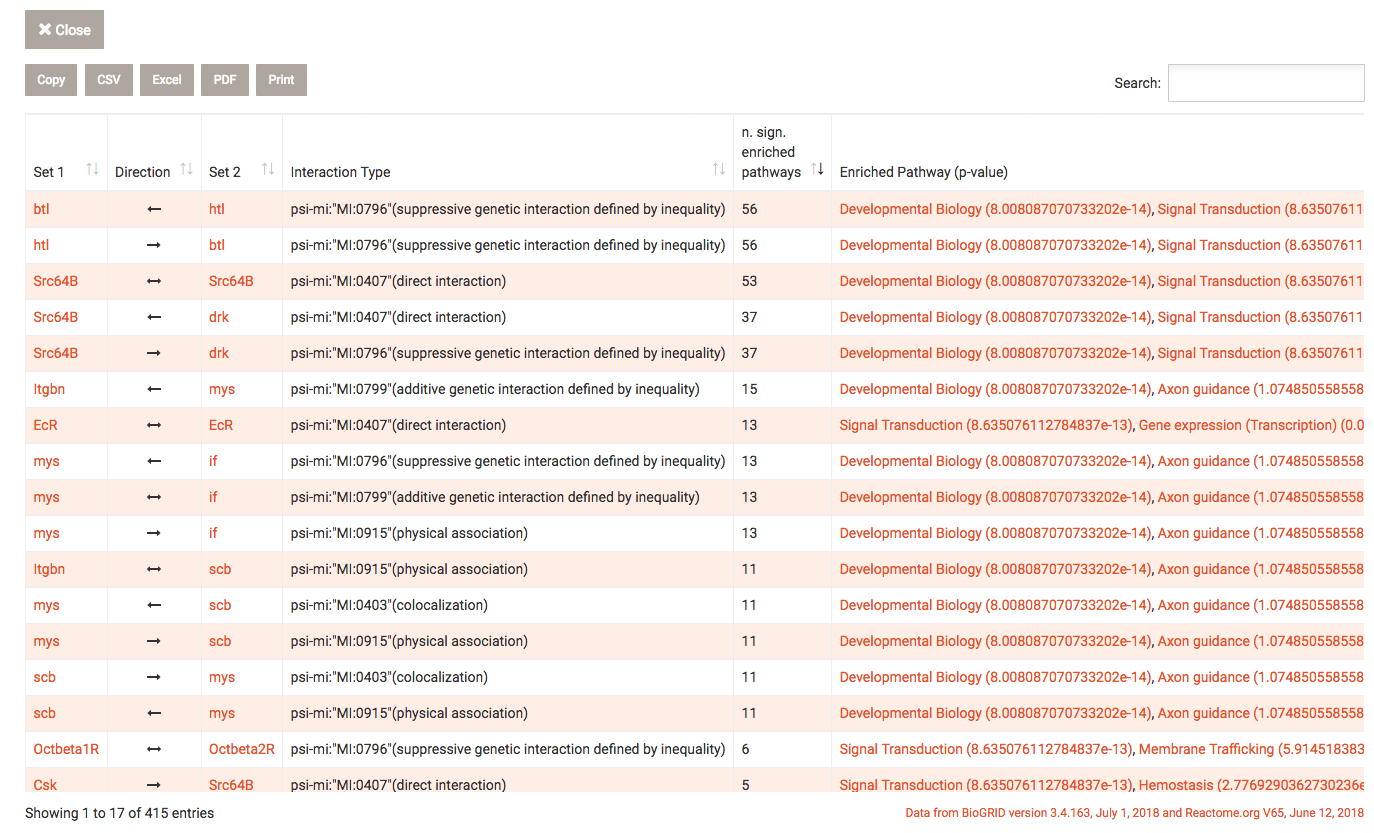
Find pairwise interactions button after selecting two analyses in the history, this overlay is shown. BioGRID
interaction data is used to find interactions between genes or gene products where one of the interaction partners is
present in the first analysis and the other in the second analysis. WEADE filters these results for genes that include
either the GO term "ligand" or "receptor".
Using Reactome.org data, WEADE tries to determine to which pathway this interaction may belong to. The genes of the
two data sets are then tested for enrichment in the pathways. Significantly enriched pathways are reported next to the
interaction.
To exit from the overlay, click the close button.
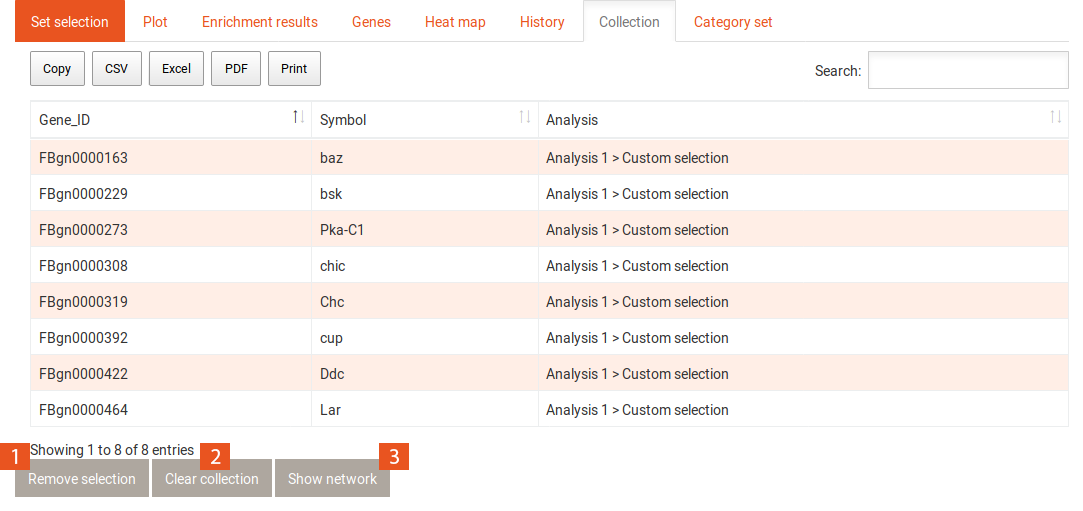
The collection shows a table of genes that have been added to it from the Genes or the Enrichment results tab. The gene identifier and the gene symbol are shown for each gene, as well as from where the gene has been added to the collection.
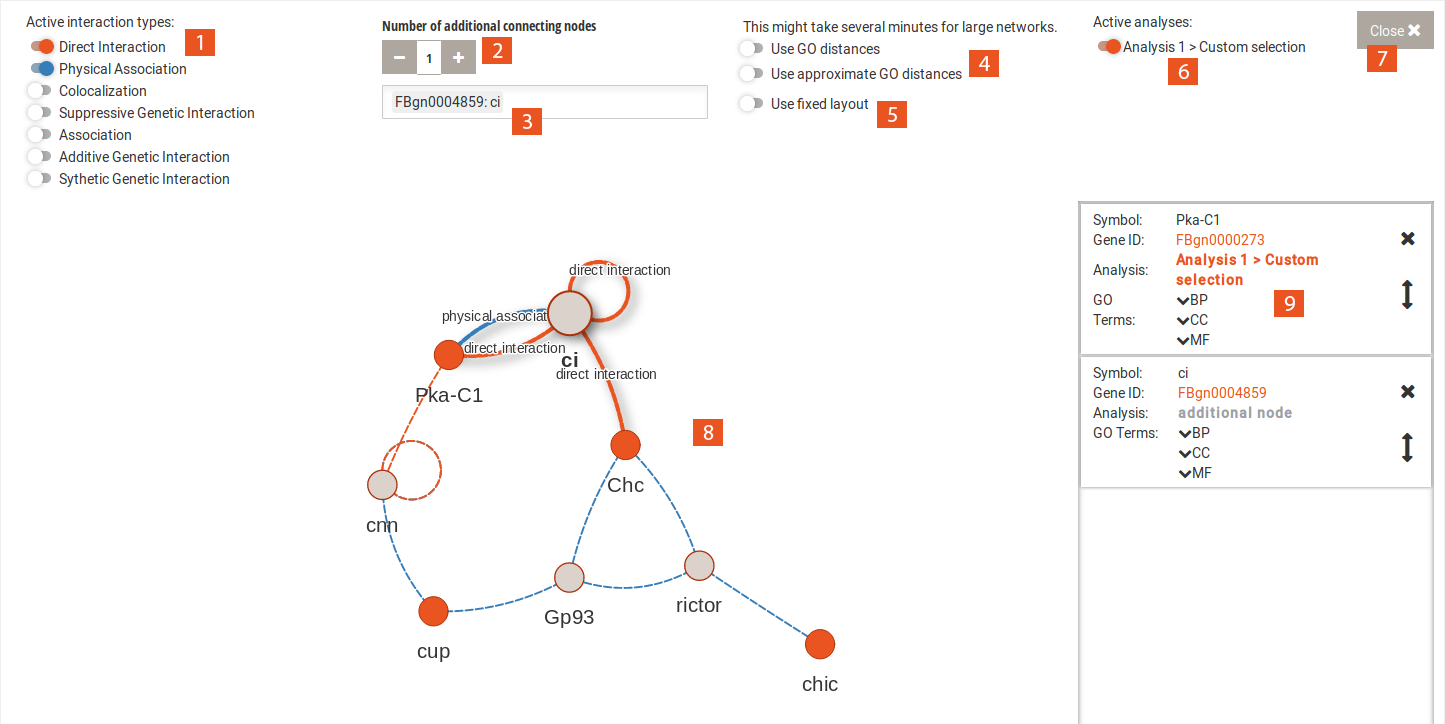
The interaction network uses BioGRID data to find interactions between genes and gene products of the selection.
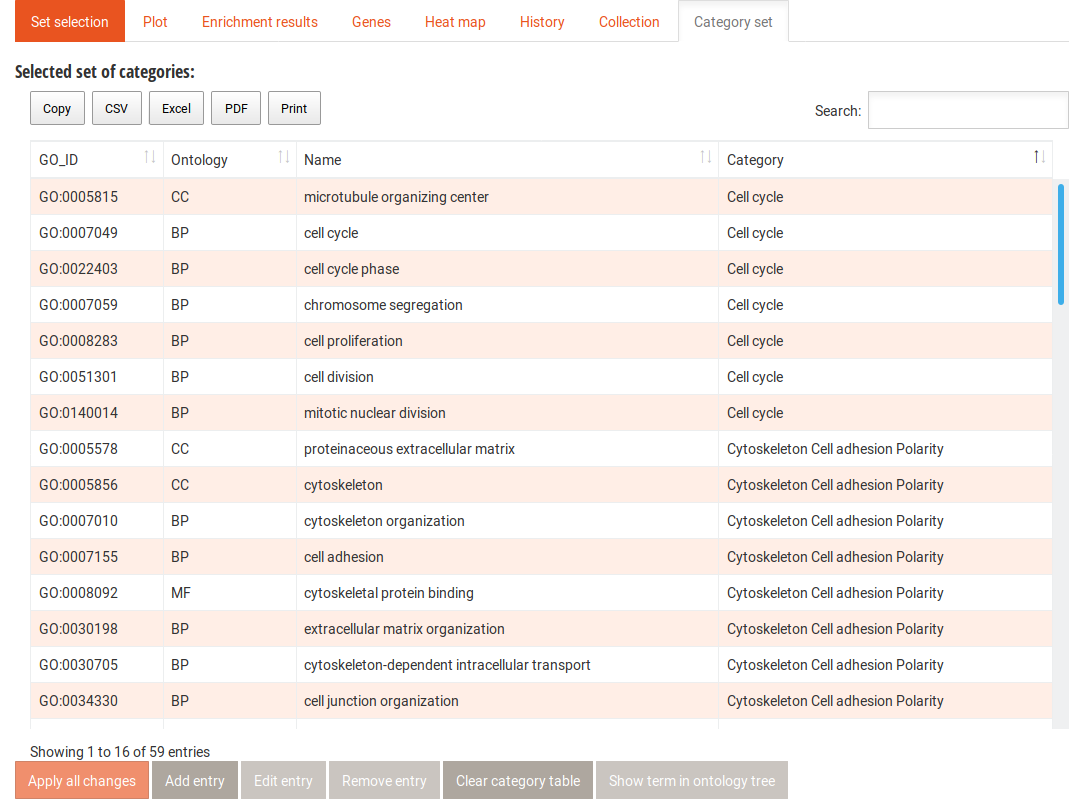
In this table, the GO terms that contribute to each category are listed. From here you can add terms to any of the categories, add new categories, and remove terms. You can also show the terms in their context in the Gene Ontology graph. After editing the category set, you can click the Apply all changes button. This creates a new category set in the drop-down menu in the Categories side tab for the duration of the session. It will automatically be selected for your next analyses.